提取拷贝数变异基本思路
以Canvas工具为例阐述提取拷贝数变异的基本思路。
讨论点:
-
对比浅全基因组测序和深全基因组测序的技术区别
-
阐述种系变异和体细胞变异提取的技术区别
首先简单介绍一下Canvas工具,该工具适用于不同测序平台测序数据的拷贝数提取,包括体细胞变异和种系变异,有四个模块:
-
Germline-WGS:通过全基因组测序提取二倍体种系样本中的CNV
-
Somatic-enrichment:通过靶向测序数据提取体细胞样本中的CNV
-
Somatic-WGS:全基因组测序数据中提取体细胞样本中的CNV
-
Tumor-normal-enrichment:靶向测序正常和肿瘤配对样本提取CNV(全基因组的情况下不设置筛选区域即可)
除此之外该工具还可以推断其他参数比如:肿瘤倍性,纯度,以及异质性。
1. CanvasBin
该步骤创建基因组窗口(windows/bins),并统计落在每个窗口内的观测到的比对序列(observed alignment)的数量。
- 推荐输入为测序深度至少为10X的bam文件(还支持kmer.fa,下面会讨论),因为需要计算等位基因频率。该步骤读取BAM文件并应用多个筛选条件:
- 只保留与正链(forward strand)匹配(proper pairs) 的读段(reads)【我的理解:这里proper pairs的意思应该是正确定向的片段,读段相对于彼此方向正确,即一个配对到正向链,一个配对到反向链】
- CIGAR字符串【点击跳转】 至少满足起始有35个的匹配(matches)
该工具把观察到的比对序列(alignment)存储在RAM中,使用每个序列的最左边碱基作为索引,分别存储在字节数组 \( O_i \) 中,对应每个染色体i。
- 除了BAM之外,输入文件还可以是kerm.fa的FASTA文件,可通过Canvas distribution得到,该文件中包含了用于序列比对的基因组序列,FASTA文件中每个35-mer【点击跳转】 的初始碱基被标记为特殊(大写)或非特殊(小写)。如果一个35-mer是特殊的,那么它的首字母就是大写的。举个例子:
acgtttaATgacgatGaacgatcagctaagaatacgacaatatcagacaa
那么特殊的35-mer就是:
ATGACGATGAACGATCAGCTAAGAATACGACAATA
TGACGATGAACGATCAGCTAAGAATACGACAATAT
GAACGATCAGCTAAGAATACGACAATATCAGACAA
特殊的35-mer在基因组上的位置存储在位数组 \( E_i \) 中,特殊的被标记为1,非特殊的标记为0。FlagUniqueKmers工具可为参考FASTA文件生成此注释。可以选择屏蔽一些特殊的35-mer子集,在命令行中提供一个BED文件,指定要忽略的基因组区域(-f,-filter=VALUE)。默认的屏蔽文件是改工具提供的fiter13.bed,该文件包含了着丝粒和端粒,以及一些中心体周围的区域。
- CanvasBin中另一个重要的输入是参数bindepth,设置二倍体区域中每个窗口预期的平均比对序列数量(desired average number of alignment per bin)(参数-d,–bindepth=VALUE,默认设置为每个窗口内有100个比对序列,和aCGH array的信噪比大致相同),然后将此值转换为“bin size”。注意,在其他的工具中,“bin size”是用来设置窗口大小的,但在这里它指代每个窗口中特殊的35-mer的数量。由于人类基因组的某些区域重复性很高,因此每个窗口的物理大小(在基因组的坐标)不相同。对每个常染色体计算观察到的比对序列数量比上特殊的35-mer数量。然后,将每个bin的期望比对数除以这些比率的中值以获得bin size。

全基因组测序数据中,bin size通常在800~1000范围内,相应的,大多数物理窗口的大小将在1~1.2kb的范围内。相比使用固定的基因组间隔,这种方法的优势在于,每个窗口里都会有几乎相同数量的读段(reads),而不用考虑窗口的“映射性”(mappability)或“唯一性”(uniqueness)。
-
CanvasBin还可以添加Nextera manifest文件(-t,–manifest=VALUE),在这种情况下,只会使用manifest中指定的目标区域来计算bin size,比如覆盖到靶向区域的(on-target region)窗口(满足desired median number of alignment)。
-
CanvasBin还允许在BED文件中自定义窗口(通过-n参数设置)。例如,可以使用探针区域作为窗口。
-
CanvasBin的输出是G1_P1_0.binned,本质是一个压缩的BED文件,包含染色体,起始和终点位置,观察到的计数值,每个窗口的GC含量,内容如下:
chr1 754148 755200 90 46
chr1 755200 756030 93 54
chr1 756030 756783 90 54
chr1 756783 757489 89 53
chr1 757489 758242 80 55
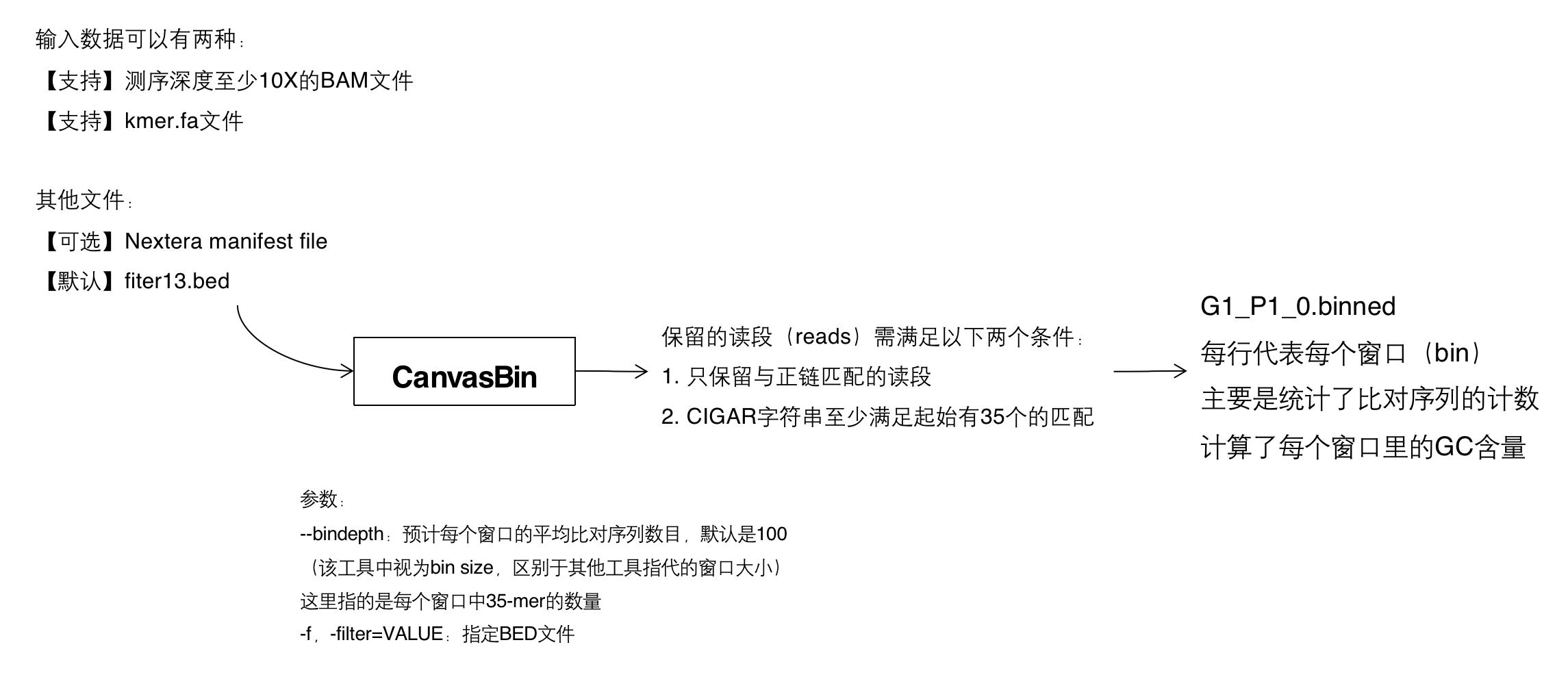
CanvasBin流程汇总
2. CanvasClean
该步骤对CanvasBin得到的结果去除离群值和系统偏差。
一共包含4个步骤:
-
单点离群值移除
-
物理大小离群值移除
-
GC含量矫正
-
福尔马林固定,石蜡包埋的样本的标准化(只用在体细胞流程中)
输入数据是CanvasBin输出的BED文件。
2.1 Single point outlier removal
定义两个bin计数:a和b,如果它们的差异大于我们在偶然情况下所预期的差异,(假设 a 和 b 来自相同的潜在分布),我们使用卡方检验统计量:

X² 超过6.635(自由度为1的卡方分布的第99百分位数),该数值可以作为排除窗口区间的阈值。
2.2 Physical size outlier removal
因为Canvas的流程中窗口没有相同的物理(指的是基因组)大小,在全基因组测序数据中他们的平均值大约是默认的1kb。然而,有些窗口可能达到Mb的大小,因为他们覆盖了基因组上的重复区域,这种高度重复区域在该步骤中被移除。计算观察的物理大小的第98百分位数,移除大于这个阈值的窗口。
2.3 GC content correction
窗口计数(bin counts)的重要影响因素之一就是GC含量。该工具提供了矫正GC含量偏差的机制:
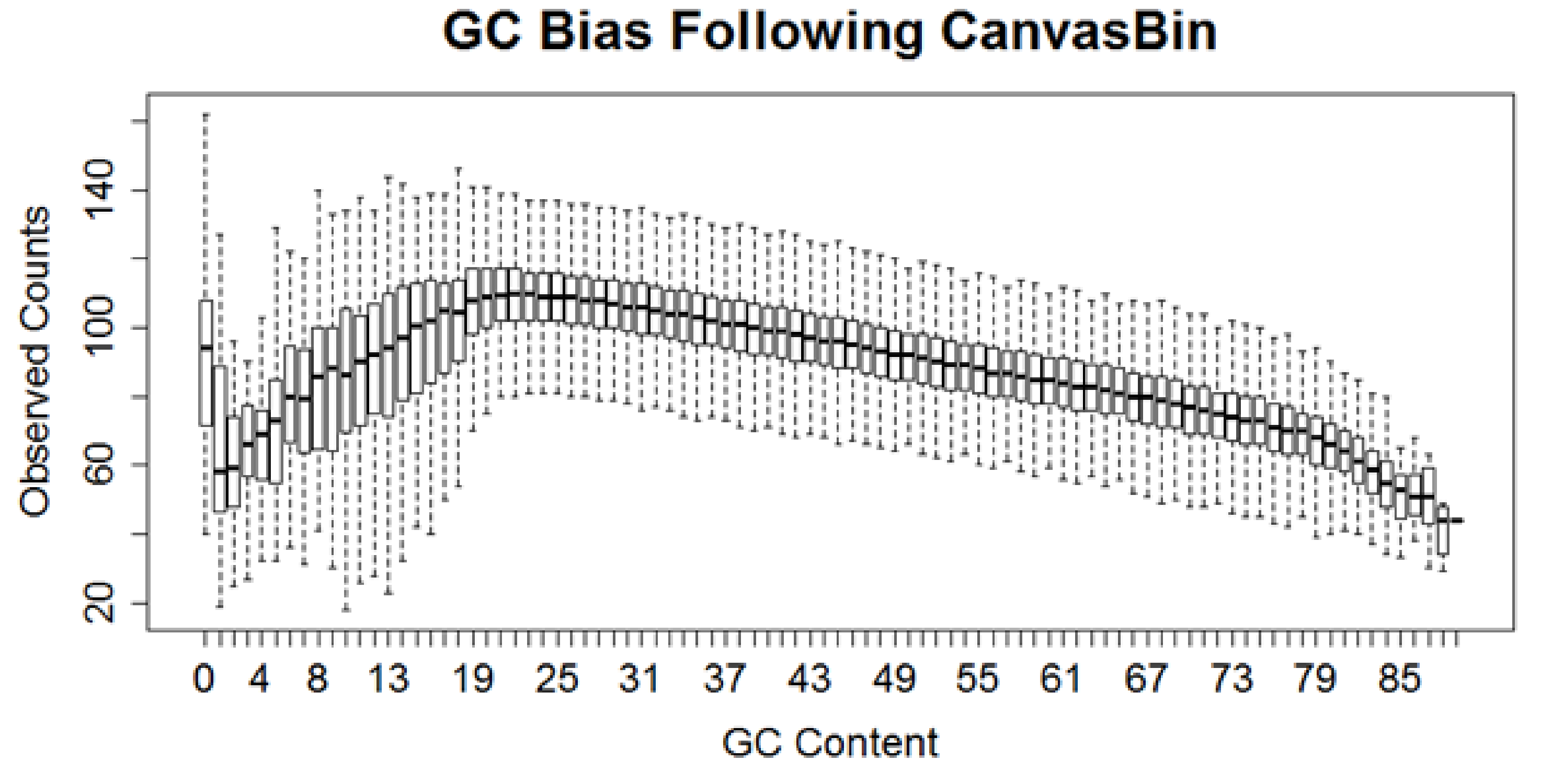
Exmple of bias
矫正是基于平均的中位数,首先把每个窗口的GC含量四舍五入到最近的整数,把GC含量相同的放在一起,在具有相同GC含量的窗口内,每个窗口的计数除以窗口的中位计数值。最后,这些数值再乘上总体的中位数。这一步的目的是使上面显示的箱线图中的箱体中点变平。
-
小于给定阈值:有些GC含量下只有很少的窗口,这样会降低中位估计的置信度,为防止这种情况发生,CanvasClean舍弃了窗口数量低于给定阈值的所有窗口(max(minNumberOfBinsPerGCForWeightedMedian, number of bins/100))。
-
小于100:如果一个GC值下少于100个窗口,使用相邻GC值的窗口来计算加权中位数。目标GC值的窗口具有全权重,两个相邻GC值的窗口具有一半权重,相隔两个窗口的具有1/4权重,依此类推。相邻的GC值将被包括在内,直到我们至少有100个b窗口来估算加权中位数的窗口数量。可以使用命令行参数 -w
来设置参数:minNumberOfBinsPerGCForWeightedMedian。
对于外显子测序数据中,CanvasClean使用Nextera manifest file(-t,–manifest=VALUE),这种情况下只有on-target的窗口才会被用来估计每个GC下的窗口的计数估计。和全基因组一样,canvas会丢弃小于阈值的窗口,在某GC含量下少于100个窗口时计算权重中位数。
在有些情况下,GC含量的矫正有非常重要的作用。比如在低深度测序的实验中使用nextera建库。
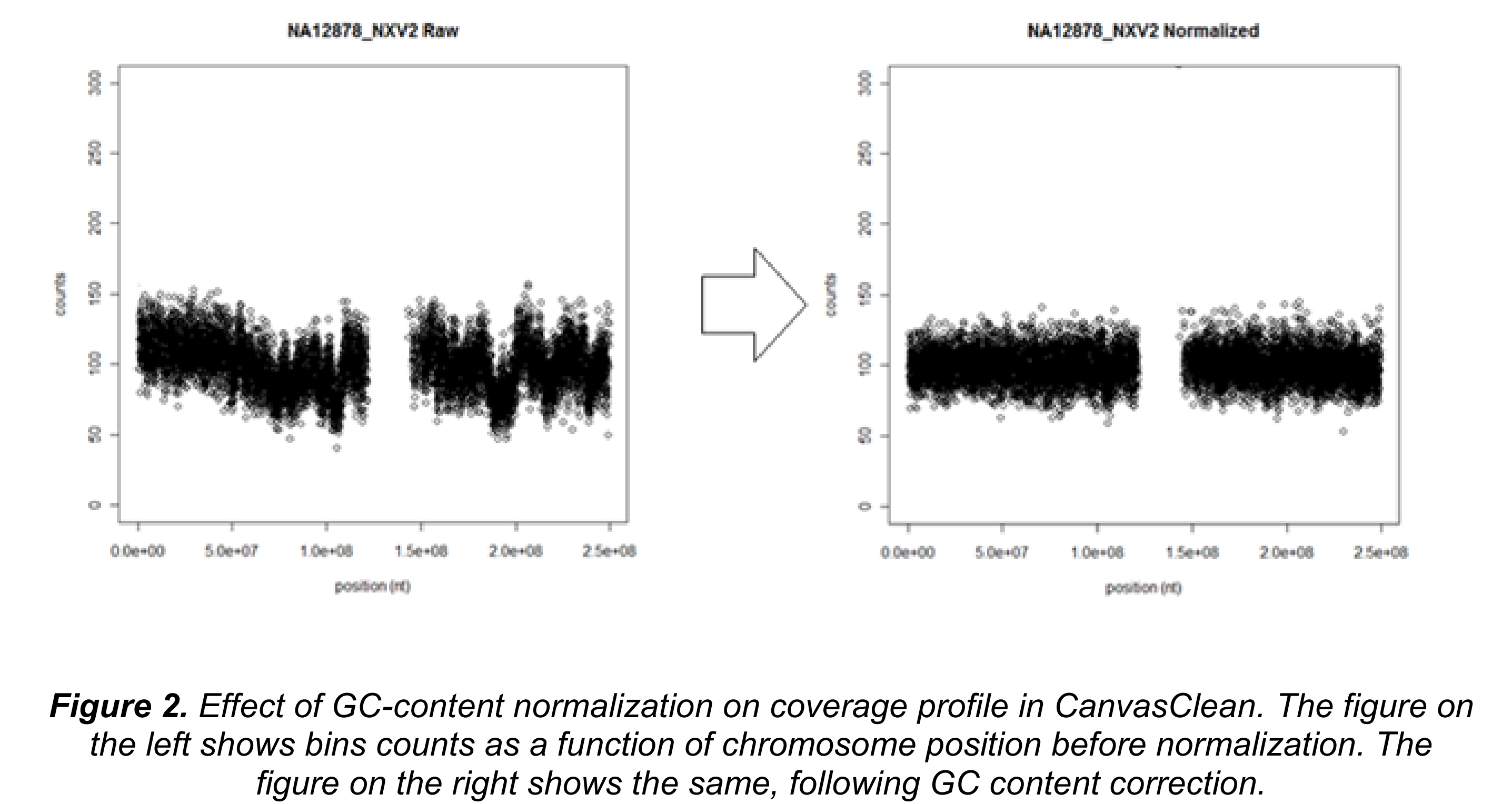
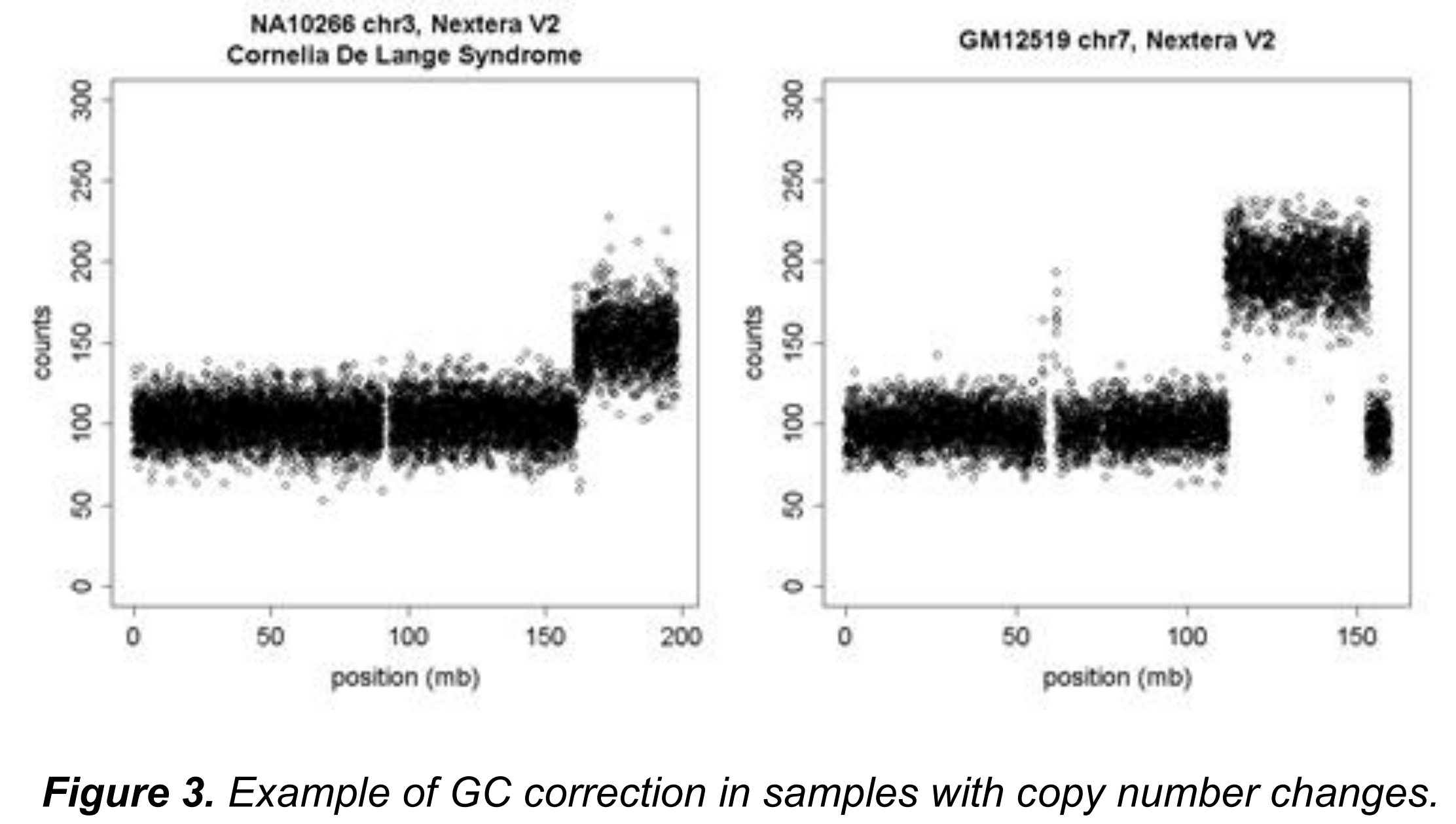
对于全基因组测序数据,通常会看到中位绝对误差(median absolute deviations, MADs)是10.3,这非常接近泊松模型预测的平均计数是100情况下MAD为10的情况,这表示完成CanvasClean步骤后误差非常微小。更重要的是,归一化并没有弱化CNV的信号。见下图:
2.4 Normalize for FFPE
福尔马林固定石蜡包埋的样本(FFPE)由于对样本的固定操作会引入噪音和偏差(比如DNA片段化问题,以及需要更高的温度进行DNA提取),导致难以解释深度异常现象。尽管其中一部分可以追溯到AT富集测序片段的丢失(部分通过GC标准化进行校正),但这并不能完全解释偏差,还存在其他尚不太清楚的因素,如染色质构象,可能也参与其中。Canvas采用两步策略来标准化FFPE样本:
-
FFPE去噪算法来移除局部偏差
-
以片段化为基础的标准化
2.4.1 FFPE去噪算法来移除局部偏差
去除FFPE特色的噪音和方差。FFPE引起的噪音主要特征是在高偏差区域的覆盖差异增加,这种偏差不能通过依赖均值和其他统计数据(中位数、众数等)的规范化操作来纠正。可以通过以下步骤来处理:

(1)计算连续窗口之间的覆盖度差异(即连续覆盖区域之间的覆盖深度的差异,这里的1可能指的是1个窗口,不确定),时间滞后差异显示了FFPE偏差区域中不属于拷贝数变异导致的方差峰值。该数据的特征有以下几个优点:
-
所有区域的均值为零(包括大范围的拷贝数变化);
-
方差是原始数据的两倍,可以使噪声区域更容易检测;
-
可以通过拷贝数变化之间更锐利的过渡来区分正确的断点区域,而不是从混乱区域中判断。
【如何区别方差的区别是由于拷贝数变化而不是由于噪音呢】
(2)这种峰值可以用于一个简单的局部FFPE偏差/方差去噪算法,CanvasClean运行一个滑动区间(默认为20个覆盖窗口),计算每个窗口内的标准差(SD),然后为每个样本估算在所有窗口和染色体上的平均标准差;为简单起见,将这样的度量称为局部标准差(localSD),在CanvasClean步骤中会移除达到局部标准差阈值的区间。由于这是一种有损转换,所以只针对噪音很多的FFPE样本,而正常和噪音较少的FFPE样本不受影响(如何判断是否需要第2步见下面的第3步)。
(3)从参考数据中针对同一个人FFPE和新鲜冻存(FF)样本的覆盖数据进行比较,局部标准差(localSD)显示,通过该度量阈值的设置(从图5中的示例中该阈值设为10)清晰地区分了FFPE和FF组,可见FF组的SD都小于10。该特征可以用来区分是FFPE样本是否需要进行去噪处理。
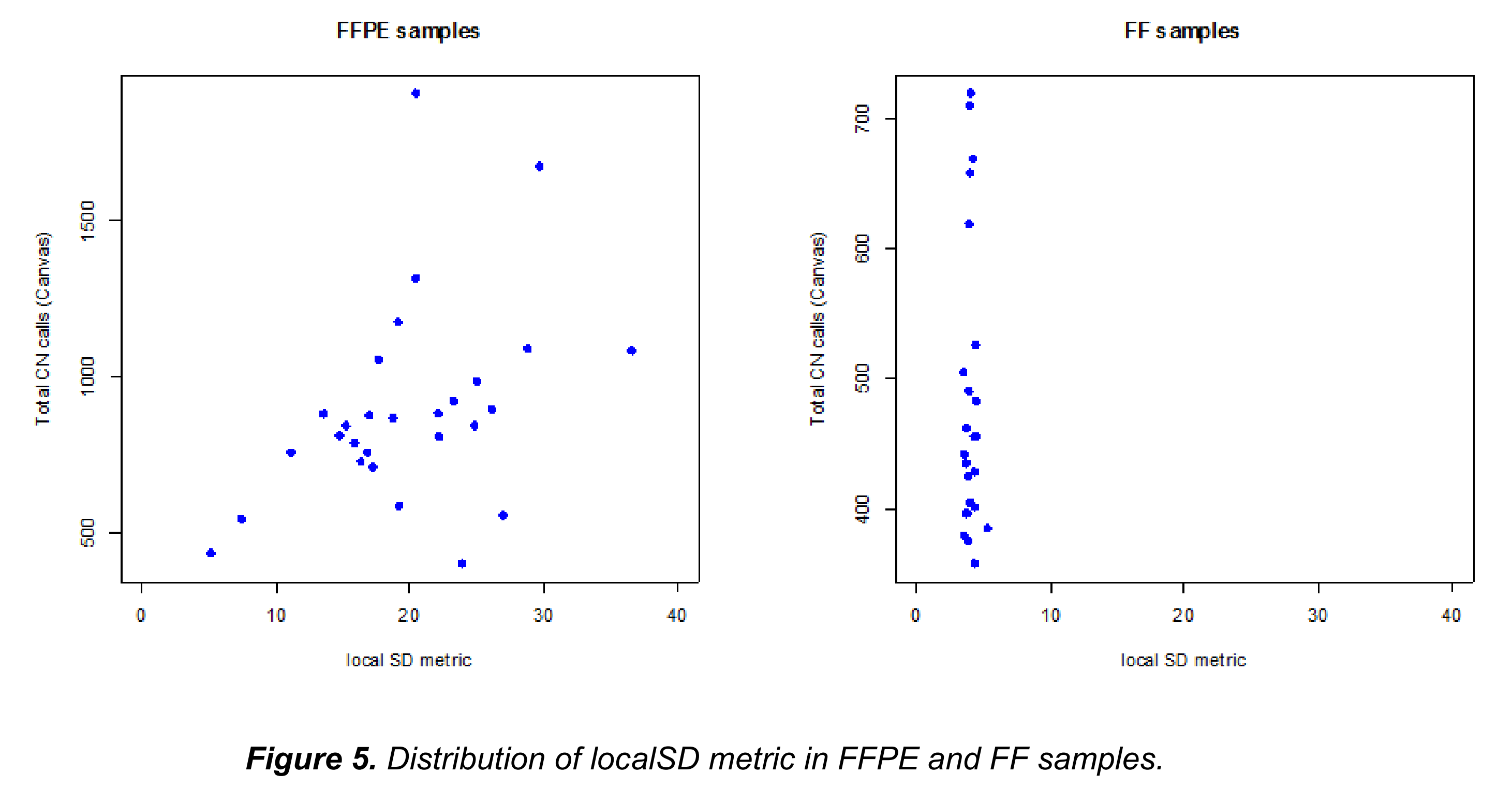
2.4.2 以片段为基础的标准化
虽然基于窗口的归一化可以纠正大部分的噪音,但仍然无法捕捉到在单个片段(fragment)大小水平的噪音。后者特别受到在FFPE文库制备过程中发生的PCR扩增步骤的影响。为了改进这一点,Canvas通过加权的GC含量校正因子来对区段计数进行标准化,该校正因子是通过对各个片段对总窗口计数的贡献进行平均得出的。片段的平均值是根据给定片段序列的观察值与期望的GC含量计数比例进行加权(图6)。片段基于的归一化可以通过在Canvas可执行命令行中指定–custom-parameters=CanvasBin,–mode=GCContentWeighted参数来启用。
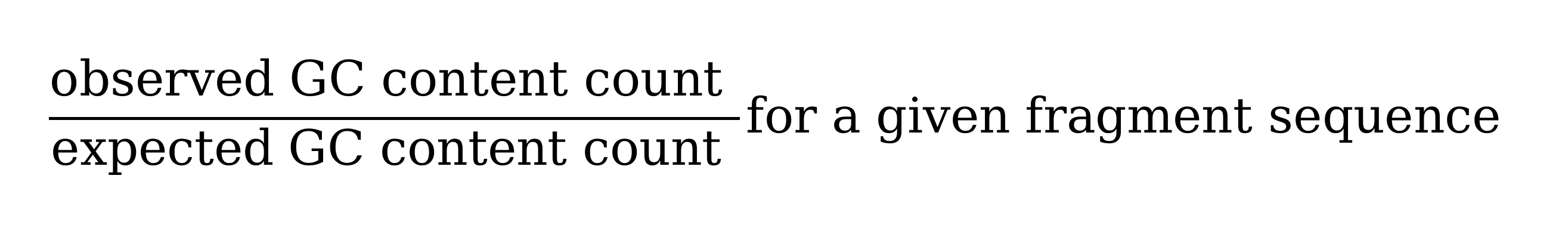
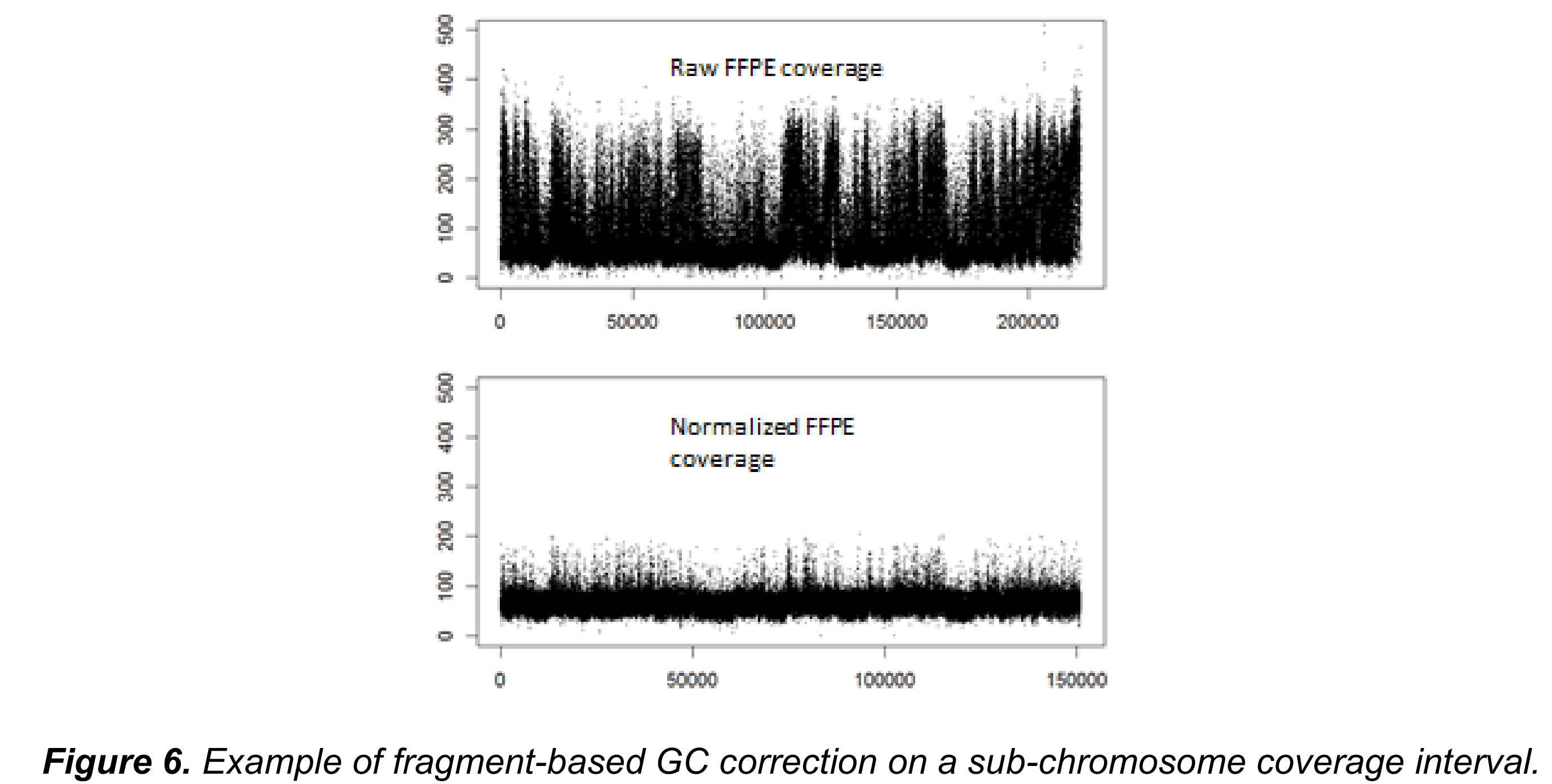
CanvasClean的输出和CanvasBin的输出格式是一样的。
3. CanvasPartition
该步骤对于标准化过的数据,把每个窗口(bin)根据相等的平均覆盖度(mean coverage)划分成不同的片段(segment)。
把标准化之后的BED文件作为输入,并将所有的窗口划分为一组具有相等平均覆盖度的不同片段。CanvasPartition实现了多种分段算法,包括 unbalanced Haar wavelets(默认)和循环二分法分割(CBS) 。
算法内容此处不做赘述,详细解释分别参考超链接。
4. CanvasSNV
该步骤用来估计SNV等位基因频率(又称最小等位基因频率,MAF)。
CanvasSNV 步骤用于估计 SNV 等位基因频率(也称为次等位基因频率,MAF=minor allele frequency),输入文件是:
-
包含变异记录的VCF文件,或正常的参考样本,或dbSNP参考VCF文件的路径
-
肿瘤和正常样本匹配的,包含比对序列读段(reads)的BAM文件
在肿瘤/正常样本分析流程中,正常样本中得到的种系SNV用来鉴定肿瘤中所有通过筛选的(FILTER一列中为PASS)杂合性SNV。(GT=0/1或1/0),另外如果GQX tag存在(变异位点的经验校准变异质量得分),需要选择该数值大于30的数据。提取种系变异时,CanvasSNV使用种系SNV或者遍历所有的dpSNP位点,对于每个SNV位点,Canvas都会计算REF和ALT等位基因的序列比对数目。这个计算过程中去除PCR重复,次要对准(secondary alignment)和补充对准(supplementary alignment)。
PS:次要对准通常指的是在DNA测序过程中,当测序仪器无法确定一个特定位置的碱基时,会有多个可能的对准方式。补充对准则是指在进行DNA序列分析时,将次要对准或低质量的序列数据与参考基因组或DNA序列进行比对,以提高数据质量和准确性。
设A为ref allele的read count,B 为alt allele的read count,MAF=min(A,B)/(A+B),MAF在二倍体区域约为0.5(实际情况下,二倍体的MAF会更低,随着测序深度的增加会接近于0.5),CanvasSNV的输出是临时文件VFResultsG1_P1.txt.gz,示例如下:
#Chromosome Position Ref Alt CountRef CountAlt
chr1 12783 G A 53 135
chr1 13116 T G 24 70
chr1 13118 A G 25 69
chr1 14907 A G 73 102
chr1 14930 A G 48 98
chr1 15190 G A 160 13
chr1 16298 C T 18 54
5. CanvasDiploidCaller
提取种系拷贝数变异的最后一步是根据CanvasPartition得到每个区域的拷贝数状态,再确定变异的最大拷贝数(copy number of major allele)。
该步骤的输入数据是CanvasPartition得到的每个片段(segment)的覆盖度和MAF数值。
提取种系拷贝数变异的最后一步,是推测Canvaspartition得到的每个区域的拷贝数状态,并且确定拷贝数变异区域的最大拷贝数(MCC,copy number of major allele),这一步骤的输入是覆盖度(coverage)和每个片段(each CanvasPartition segment)的MAF数值,使用具有每个拷贝数和MCC状态的单独分量的高斯混合模型,例如对于CN=2模型有两个状态,分别表示二倍体(MCC=1,CN=2)和LOH(MCC=2,CN=2)。均值和协方差矩阵是从二倍体模型的数据中估计出来,然后通过期望最大化(EM)算法进行调整。例如,如果在二倍体模型中估计的均值µ和标准差σ为100和15,那么在单倍体模型中的对应估计值将初始化为µ/2和σ/2。在进行EM拟合后,将使用具有最高后验概率的[CN,MCC]模型元组来为每个片段分配拷贝数。在片段没有跨越SNPs(单核苷酸多态性位点)且因此MAF值为空的情况下,不考虑LOH状态。
这个步骤中具有相同拷贝数的临近区域合并为一个片段。
最终,类似于Phred的q分数被分配给每个区域。为了得出这些分数,在一个包含约10个筛选过的种系样本的测试语料库上训练了一个 logistic 回归模型,以计算每个片段具有正确方向的概率,即删除、扩增或无变化。模型使用的三个特征是:
-
LogBinCountlog10(1+bin的数量)。拥有更多bin的分段可以更准确地分配拷贝数。
-
ModelDistance:观察到的覆盖度和MAF值与所选模型预测值之间的距离。较低的距离表示拟合更好。
-
DistanceRatio:最佳模型点的模型距离与次优模型距离之间的比率。这个值越大,分配的多倍体数是正确的可信度就越大。
然后,概率被缩放以生成Phred-scaled似然度。非二倍体区域的坐标及其q分数被写入VCF文件。在为变异分配PASS标志时应用了两个过滤条件,首先,变异必须具有10或更高的q分数。其次,变异的大小必须为10kb或更大。
6. CanvasSomaticCaller
该步骤用于肿瘤和正常样本匹配的情况,鉴定拷贝数变异区域。
该步骤用于提取拷贝数变异的区域(比如:常染色体上的拷贝数不是2,或者b等位基因频率不是50%左右的区域)来估计样本纯度或者肿瘤倍性。
6.1 体细胞模型
设计原理:
- 对于给定的倍性和纯度,可以对每个拷贝数状态计算预期覆盖值和MAF。区域中给定倍性(拷贝数和最大等位基因计数),每个SNV的最小等位基因频率理论上符合:
Ploidy A: MAF 0
Ploidy AB (normal): MAF 0.5
Ploidy AA (copy-neutral LOH): MAF 0
Ploidy AAB: MAF 0.33333
Ploidy AAA: MAF 0
Ploidy AABB: MAF 0.5
Ploidy AAAB: MAF 0.25
Ploidy AAAA: MAF 0
(etc.)
-
模型的偏差(deviation) = 预期(expected)覆盖度(coverage)-观察覆盖度
-
两个模型具有相似的偏差时,偏向选择具有更小拷贝数的模型(比如,更接近二倍体)。
-
训练样本中具有已知的倍性和纯度数值,用机器学习来推断用来区分正确和错误的体细胞模型的最佳特征。
如果肿瘤是多克隆的,该步骤还会估计整体的变异杂合性,对燃预测的倍性和纯度和拷贝数状态是线性关联的,但是杂合性会导致模型更复杂。该步骤使用了很多近似的方法来检测克隆。
流程:
-
输入CanvasPartition得到覆盖片段(coverage segment)
-
输入CanvasSNV得到的变异频率(variant frequency),保留所有ref+alt覆盖度大于10的位点,如果使用了匹配的正常样本,那么CanvasSNV输出就只保留了杂合性SNV的变异频率,但是,dbSNP VCF可以用来代替正常的VCF,这样的话,CanvasSNV里面并不是所有的位点都是杂合的,这种情况可以可以调用参数-d(–dbsnpvcf)来对每个片段(segment)调整纯合位点的MAF。MAF选择的标准如下:
-
CanvasSomaticCaller会在以下情况下过滤掉片段中的零MAF(最小等位基因频率):如果超过10%的MAF是非零的,或者至少有一个位置的MAF >= 0.2。这是因为预期纯合位点的MAF应为零。
-
如果在过滤之前的MAFs数量大于给定的MAF计数阈值(默认为50),但在过滤后小于此阈值,则阈值将降低为过滤后的MAFs数量。
- 对于CanvasPartition得到的每个片段(segment),计算每个窗口(bin)计数的中位数,以及中位最小等位基因频率(MAF)。
- 每个段都被分配一个权重,该权重用于计算下面所描述的偏差。如果总片段数大于100,则权重等于片段的长度。否则,权重等于片段中的窗口数。如果片段中的MAF少于10个,那么权重将乘以MAF数量除以10的比率。这是因为较少的MAF对片段的真实中位数MAF的估计不太可靠。这种段加权技术使模型拟合更加稳健,能够更好地抵抗噪声。
- 迭代计算不同的二倍体覆盖度D和纯度p来获得模型的偏差(deviation)。
-
给定一个候选元组(D,p),计算每个拷贝数值的期望(MAF,Coverage)点。
-
(D,p)的模型偏差是在所有分段上的总和,表示为 ||(SegmentMAF,SegmentCoverage) - (ModelMAF,ModelCoverage)||。
- 估算模型的惩罚项。为了计算惩罚项,对于每个模型,从初步的全基因组拷贝数调用中计算了百分比正常、百分比CN=2和二倍体距离变量。然后,它们被用作逻辑回归模型中的预测变量,该模型在一个包含58个肿瘤/正常配对的数据集上进行训练,数据手动分配了倍性和纯度值。来自这个模型的回归系数被转换成最终惩罚项的新权重,具体如下。
-
0.25*DiploidDistance:将给定基因组转化为二倍体所需的CN事件数量的比例
-
+0.35*CN2:CN = 2 的百分比
-
+0.3*PercentNormal:正常CN的百分比
-
考虑肿瘤的多克隆性。为此,使用EM算法将每个段元组(SegmentMAF,SegmentCoverage)分配给不同的聚类。聚类是通过从已根据k最近邻距离(其中k设置为总段数)排序的观察段中抽样(SegmentMAF,SegmentCoverage)值来初始化的。只使用前70%的段。这个值是为了偏好初始EM值接近与主要拷贝数变化相关联的聚类而设定的。通过使用Silhouette系数(Rousseeuw P., 1987)选择了最佳聚类数。聚类偏差(D,p)定义为属于该聚类的段与给定模型中计算的最接近的预期(MAF,Coverage)元组之间的距离之和,如第4步所计算。一般来说,距离预期(MAF,Coverage)中心最远的聚类更有可能是异质的:如果聚类偏差>总偏差*1.5,则在计算总体聚类偏差时不考虑该聚类(该值经过训练语料库优化)。
-
总偏差被定义为模型偏差、簇偏差和惩罚项的平均值。使用具有最低总偏差的覆盖度(多倍体)和纯度模型来分配拷贝数调用。
6.2 拷贝数估计
最终需要对数据进行汇总。临近的具有相同拷贝数的片段将被合并成一个片段,片段大小低于阈值(默认50000个bases)被合并成最优的片段(最高的q值)。合并需要满足条件:在同一条染色体上,之间没有其他的片段,两者中间没有被屏蔽的片段(比如BED文件中指定需要去除的区域)。对每个变异计算Phred Q值,一个逻辑回归模型已经在一个包含33个经过筛选的肿瘤/正常样本的数据集上进行了训练,以计算每个片段正确的概率。所使用的三个特征分别是:
LogBinCount:LogBinCount是log10(1+bin数)。拥有更多窗口的片段可以更准确地分配拷贝数。
ModelDistance:模型距离。较低的距离反映出更好的拟合。
DistanceRatio:距离与最佳模型点距离与次优模型点距离之比。这个值越大,表示分配的倍数确实是正确的可能性越大。然后,将概率缩放以生成类似Phred的似然性,表示结果是正确的。请注意,这个q-scoring模型使用了与germline(CanvasDiploidCaller)相同的特征,但是在肿瘤/正常数据上重新训练。
CanvasSomaticCaller的主要输出是一个VCF格式。拷贝数根据拷贝数计数和期望计数报告为REF、LOSS或GAIN。通常期望计数为两个(拷贝数2是REF,拷贝数0或1是LOSS,拷贝数3或更多是GAIN)。当传递给Canvas的ploidy BED文件时,它可以更新特定染色体的期望拷贝数。例如,对于XY样本的染色体X和染色体Y,X或Y上的拷贝数为2会被称为GAIN。
CIGAR string
CIGAR字符串是一种在生物信息学中常用的格式,用于表示比对两个生物序列的结果,通常是DNA、RNA或蛋白质序列。CIGAR代表"Compact Idiosyncratic Gapped Alignment Report",它描述了两个序列之间的匹配、插入和删除等信息。
CIGAR字符串由一系列数字和字母组成,表示了比对的特征。其中常见的一些符号包括:
- M:匹配的碱基对(match)
- I:插入到参考序列的碱基对(insertion)
- D:从参考序列中删除的碱基对(deletion)
- N:跳过参考序列的碱基对(skipped region)
- S:从比对的序列的开头或结尾剪切的碱基对(soft clipping)
- H:从比对的序列的开头或结尾硬剪切的碱基对(hard clipping)
一个典型的CIGAR字符串示例可能是:“50M2I10M”,表示有50个匹配的碱基对,然后插入了2个碱基对,最后又有10个匹配的碱基对。
CIGAR字符串是在比对算法(如BLAST、Bowtie、BWA等)的输出中常见的一部分,用于描述比对的结果,以便进一步的生物信息学分析。
kmer
就是长度为k的DNA片段,是reads的部分片段。k-mer的作用有3个,一个是用来拼接出contig,一个是用来识别错误测序,杂合等位基因和重复序列的reads,还有通过K-mer估计基因组大小及杂合度。k-mer的数量,假设100bp的reads,35-mer,得到总数为100-35+1个kmer,不同kmer的overlap的长度为 kmer的长度减1。
