CNV
一、 拷贝数变异的介绍
1.1 结构变异和拷贝数变异
结构变异(structure variation, SV)是指基因组上大片段碱基的缺失、插入、重复、倒位和易位。这部分变异的频率,和疾病或者表型的关系等等都不明确,另外还有一部分的变异也属于结构变异,比如异态性(heteromorphisms),脆性位点(fragile sites),marker染色体,等臂染色体(isochromosomes),双微体(double minutes)等等,这部分的结构不正常和疾病有关。
等臂染色体,是两条基因和形态都一致的染色体臂。
双微体,是无着丝粒的,染色体外扩增的核染色质,通常包含特定的染色体片段或基因,在癌细胞中常出现。
marker染色体,也被称为结构外异常染色体或“多余”染色体。在荧光原位杂交实验中,除了正常的染色体补体外的染色体。
拷贝数变异可以看成是特殊的结构变异。鉴定SV对DNA测序深度有较高的要求(一般要>30x),而较低深度的(>5x)覆盖均一的DNA测序就可以鉴定到CNV。对于肿瘤样本中的体细胞CNV和SV,需要比较肿瘤样本和正常样本来得到肿瘤样本特有的变异。基因芯片技术分辨率较低,能检测到的片段都是大片段的(比如1kb以上)。二代测序对于比较大片段的SV,比如是超过150bp长的变异,一个reads是无法涵盖这个变异的,这种情况下会有不同的策略进行间接地推断。
1.2 结构变异和拷贝数变异造成的影响
-
Cancer
-
精神类疾病
-
免疫类疾病(红斑狼疮)
-
21三体综合症:做缺陷新生儿产前诊断
……等等
二、拷贝数变异的检测
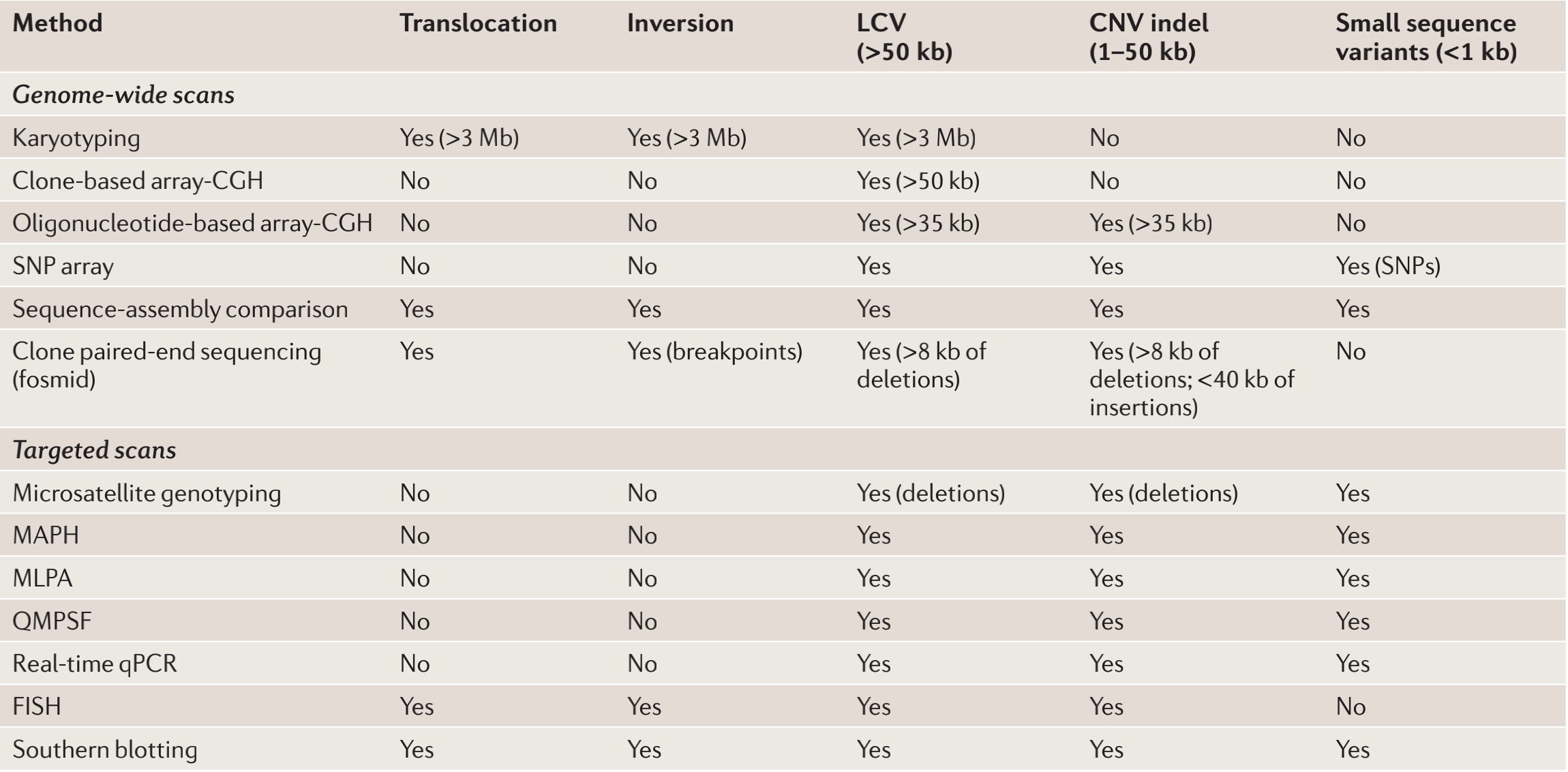
检测人类基因组中结构变异的方法
CNV, copy-number variation :拷贝数变异
CGH, comparative genome hybridization:比较基因组杂交
LCV, large-scale CNV :大范围拷贝数变异
FISH, fluorescence in situ hybridization :荧光原位杂交(
Indel, insertion and deletion:插入和删除
MAPH, multiplex amplifiable probe hybridization:多重可扩增探针杂交
MLPA, mutiplex ligation-dependent probe amplification:多重连接探针扩增技术
QMPSF, quantitaive multiplex PCR of short fluorescent fragments:短荧光片段的多重定量PCR
qPCR, quantitative PCR:定量PCR
2.1 传统技术
2.1.1 细胞遗传学中常用染色体核型分析:
- 利用PHA(植物血凝素)刺激成熟的淋巴细胞再次分裂
- 利用秋水仙素在细胞分裂中期破坏纺锤丝,抑制细胞分裂,形成染色体的形态
- 通过胰酶消化或缓冲液作用,将染色体显带,通过带纹和数目的分析判断染色体数目和结构的情况
2.1.2 荧光原位杂交(FISH)
……
2.2 aCGH芯片(array-based Comparative genomic hybridization)
2.2.1 aCGH和CGH的异同
1992年,Kallioniemi发明了无需细胞培养的比较基因组杂交技术(CGH):
- 用不同荧光分别标记肿瘤和正常对照样本,把两者DNA等量混合
- 与中期细胞染色体进行杂交
- 通过比较肿瘤和正常对照荧光信号的相对剂量来检测拷贝数异常,DNA局部扩增或缺失导致单个染色体上的荧光强度增加或减少
该方法的缺点是无法检测整个染色体组数目增加的多倍体变化,也无法检测DNA总数量不变的染色体畸变。
1995年。Schena等利用CGH技术结合微阵列基因探针(aCGH)定量检测多个基因的表达,随后aCGH应用于检测拷贝数异常。和CGH相比,aCGH的优点是:该技术不需要染色体培养,一次杂交实验即可在整条染色体或染色体区带水平对不同基因组间DNA序列拷贝数的差异进行检测并定位,aCGH将两个样本的DNA用红绿两种荧光进行标记,然后与芯片进行杂交,用据分析软件检测红绿两种荧光的比值,分析相对对照样本,实验样本的DNA拷贝数是增加还是减少。
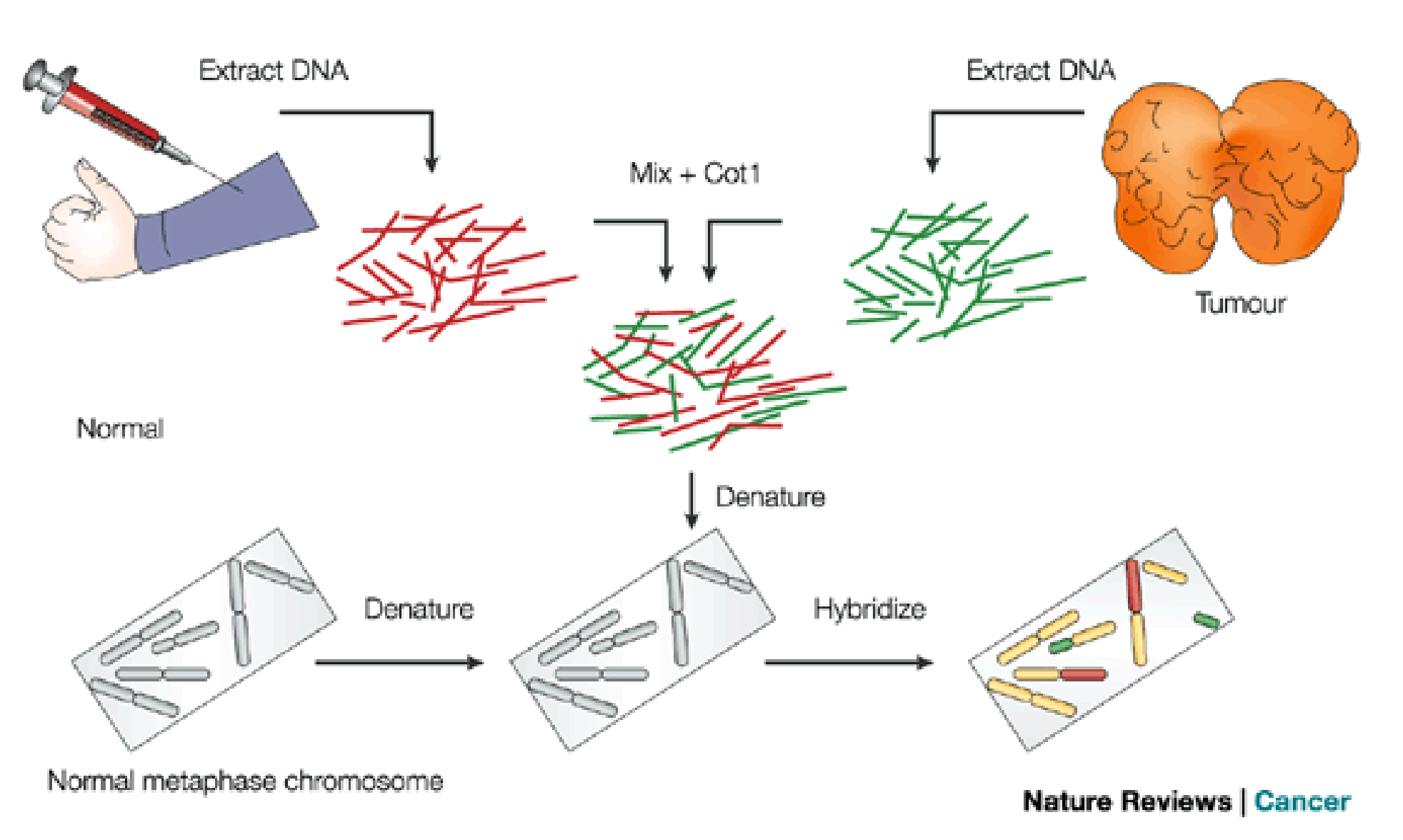
比较基因组杂交
“dye-swap"检测了假信号,该方法进行了两次检测, 在第二次检测时颠倒肿瘤和正常样本的标签,这样,如果两次检测的信号有不对称的情况时,认为是 假信号。
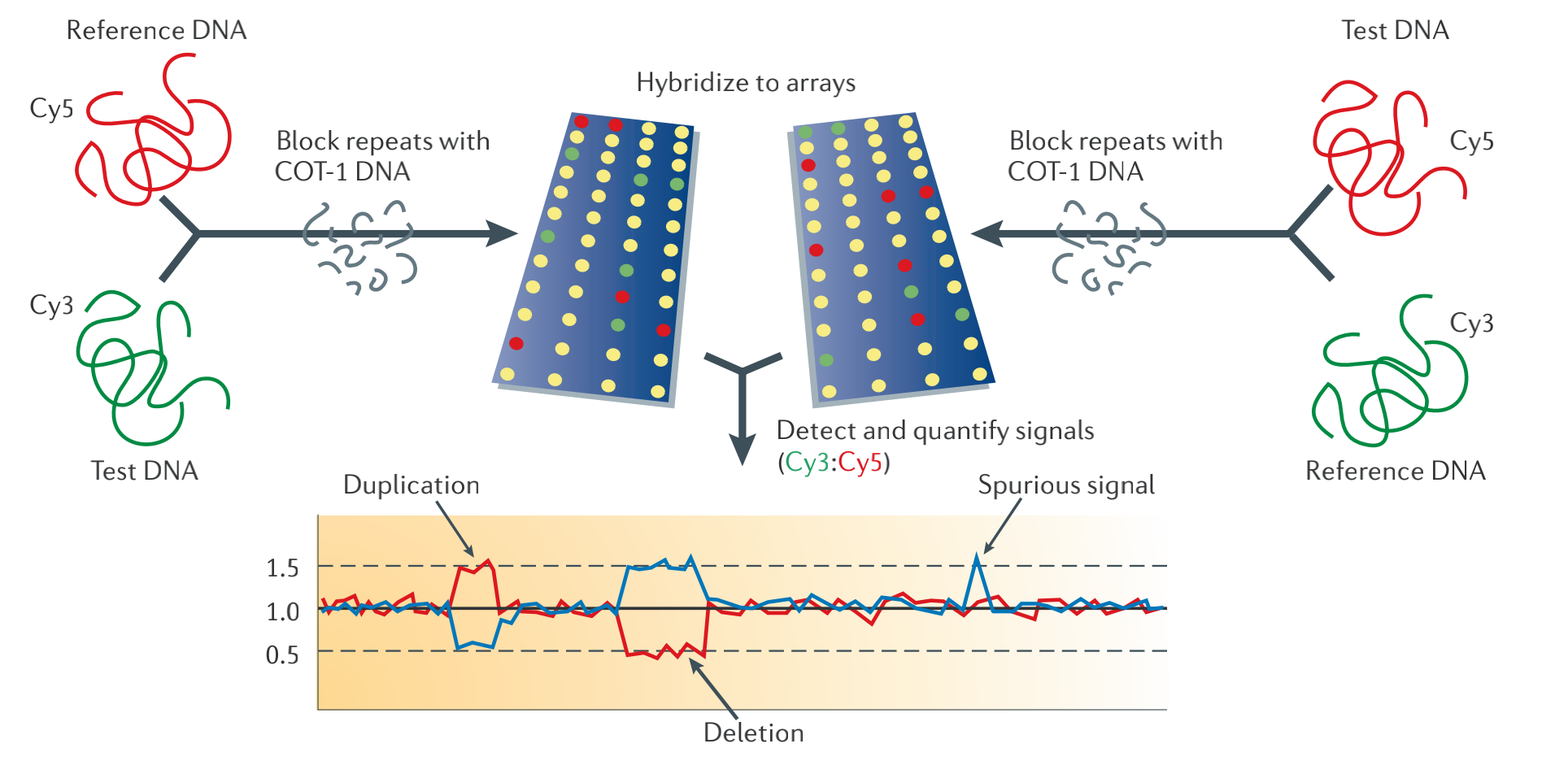
基于aCGH芯片技术的全基因组拷贝数变异检测
对于芯片技术来说,探针的本质是一段很短的DNA序列,是提前设计好的,对于倒位(inversion)和易位(translocation)来说没有改变DNA的剂量,在芯片上看不出来,另外由于插入(insertion)的片段可能是原先基因组上没有的,而探针是根据已知序列进行设计的,因此插入也无法进行检测。
2.2.2 数据拟合
软件最原始的输入数据为荧光的信号值,信号值是有波动的,而拷贝数一定是一个整数,算法通过对原始的荧光信号值进行拟合,以确定对应染色体片段的拷贝数水平。
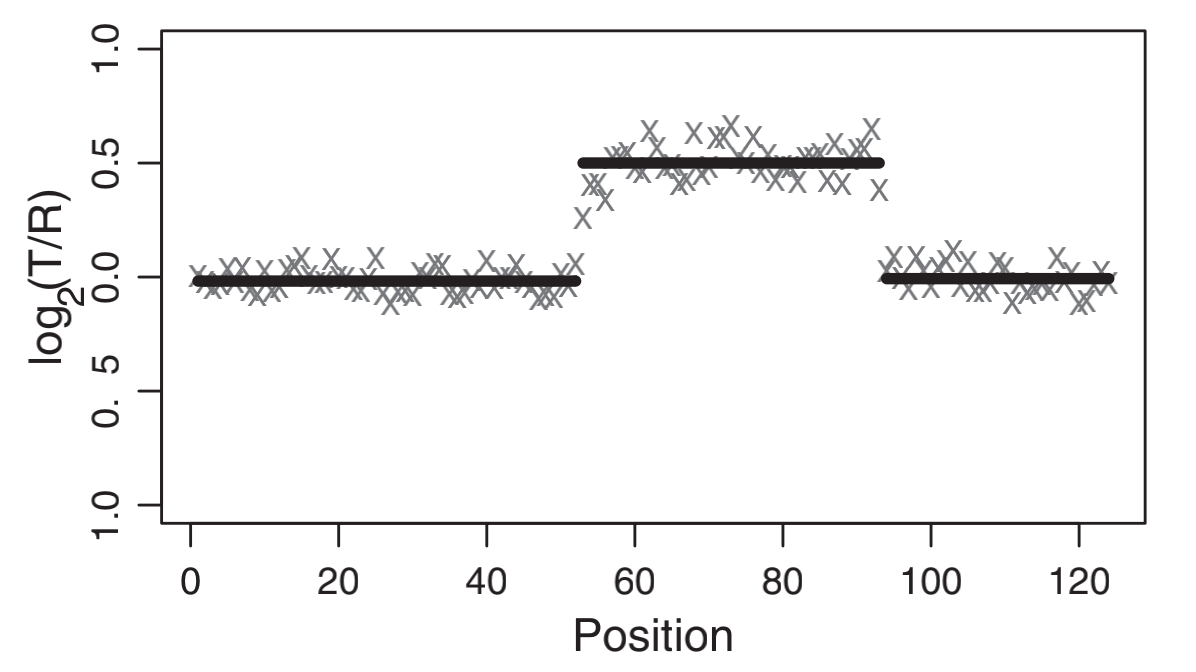
离散的拷贝数数值
- 去除噪音
- 检测正常、增加、减少的拷贝数
- 断点的检测
2.2.3 断点检测之CBS(circular binary segmentation)
是一种基于染色体芯片数据检测拷贝数变异的方法,目前也适用于二代测序数据的拷贝数变异检测的方法。
A. CBS的背景
芯片数据存在噪音,发生在连续区域的变异通常会覆盖多个标记(marker),因此标记不能够完全反映test样本中真实的拷贝数变异情况,因此需要一个方法来将染色体划分成多个片段,保证每个片段中的拷贝数是一致的。循环二元分割算法是从1975年的二元分割算法改进来的,它提供了一种自然的方法将染色体分割成相邻的区域,并利用permutation reference distribution避免了数据的参数化建模。
B. CBS算法
循环二元分割算法(circular binary segmentation, CBS)是目前常用的芯片数据分段算法, 其优势在于利用相邻待测区间的数据均值差构建 t 统计量, 进而精确检测不同变异区域间的分段点。介绍一下估计拷贝数变异区域位置与变点检测(change point detection)的关联。
原始的变点检测策略是1975年Sen和Srivastava提出来的,假设\( D \) 是芯片数据,\( n \)是长度,设\( \mu_{i} \)和\( \mu_{i}^{’} \)分别为\( D \)第一个元素\( i \)的log2ratio均值和后一个元素\( n-i \)的log2ratio均值,把\( i \)定位在使得\( \left | \mu_{i}-\mu_{i}^{’} \right | \)最大化的位置上,并且使用\( t \)检验来判断两段信号差异是否显著,如果显著,则标记此处为一个变点。原始的方法是检验1个单个变点的情况,不能够在大片段中检验出小片段的拷贝数变化,CBS算法所做的改进在于:
并非考虑单个的变点,而是假设片段环绕成一个圆圈,第一次运行时将数据划分为两个弧,从\( i \)到 \( j \)为第一个弧,从\( j \)经过\( n \)和\( 0 \)再到\( i \)为第二个弧,找到使得两段均值最大的位置,并使用\( t \)检验判断两段的均值是否显著有差异,差异显著则标记为一个变化片段(2个变点),接下来,递归地将算法应用于三个结果片段:从0到\( i \);从\( i \)到\( j \),从\( j \)到\( n \),直到发现不了新的变化片段。
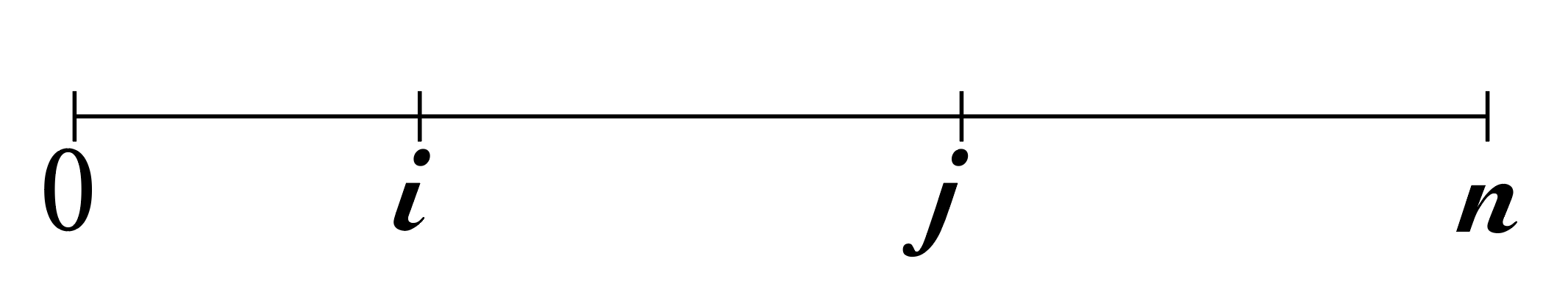
BS算法图解
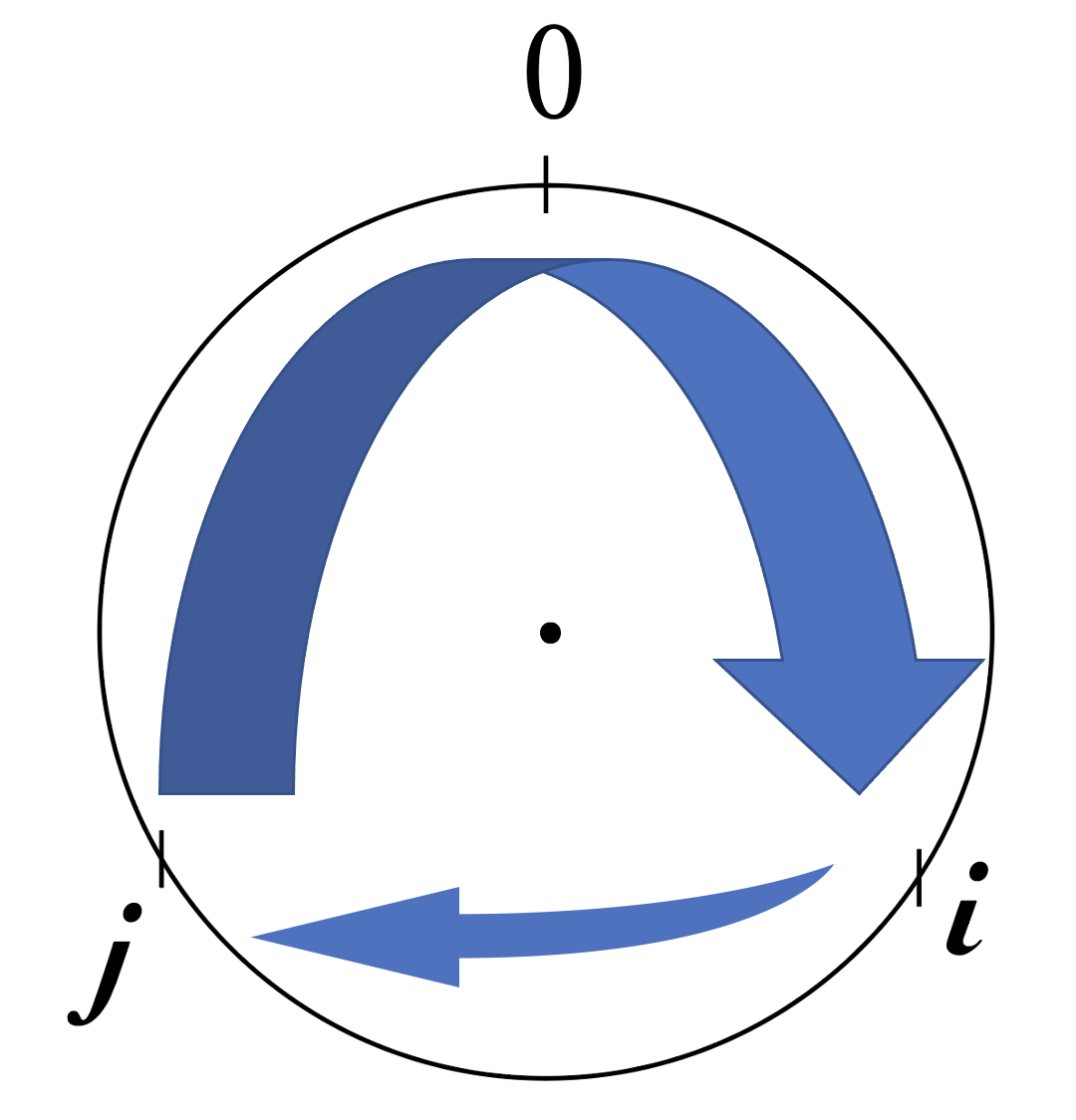
CBS算法图解
CBS算法存在的问题是存在边界效应(edge effect),即假设\( i \) 和\( j \)符合\( \left | \mu_{i}-\mu_{i}^{’} \right | \)最大化,不管是\( i \)贴近1还是\( j \)贴近n,有可能只存在一个真正的变点而非预想的两个变点的情况。可以通过以下办法来避免:
- 首先使用BS测试(CBS的前身)是否数据支持\( i \)作为一个变点,如果不是则撤销对\( i \)的标记
- 同样对\( j \)也进行BS测试,是否数据支持\( j \)作为一个变点,如果不是则撤销对\( j \)的标记
C. 使用CBS算法进行分段的工具
- R包:DNAcopy
- CNVkit:适用于全外显子,目的区域靶向测序等数据的CNV检测
- ……
2.3 SNP芯片
SNP芯片本质是基于染色体区域内的SNP分型结果来判断对应拷贝数,SNP芯片的分型是通过比较A/B两种allel对应的荧光信号强度的比值来确定的。除了提供 拷贝数信息,SNP array还提供了表型信息比如:杂合性位点丢失,证明了 删除 以及 单亲二体(Uniparental disomy, UPD)的存在。
单亲二体: 是指子代的一对同源染色体全部或者 部分 来自父亲或母亲中的一方
CCLE和TCGA里的拷贝数变异文件就是由芯片数据——Affymetrix SNP 6.0 array数据处理得到的,该芯片包括单核苷酸多态性(SNPs)和检测拷贝数变异的探针,是全基因组水平的探针,包含超过906,600 SNPs和超过946,000检测拷贝数变异的探针,和SNP 5.0 Arrays的482000 SNPs相比较,新增的424,000 SNPs是HapMap项目(在全基因组规模上,确立SNP在人群中的常见分布和传递模式)中得到的,新的标记在染色体X和Y以及线粒体SNP等表现更好。946,000个非多态性的拷贝数探针中744,000个探针是通过空间分布选择得到的,其他202,000是基于Toronto Database of Genomic Variants(DGV)已知的拷贝数变化,这样的数据可以用来从头检测拷贝数变化,以及通过对SNP和已知的拷贝数多态性位点(copy number polymorphism loci)进行关联研究。
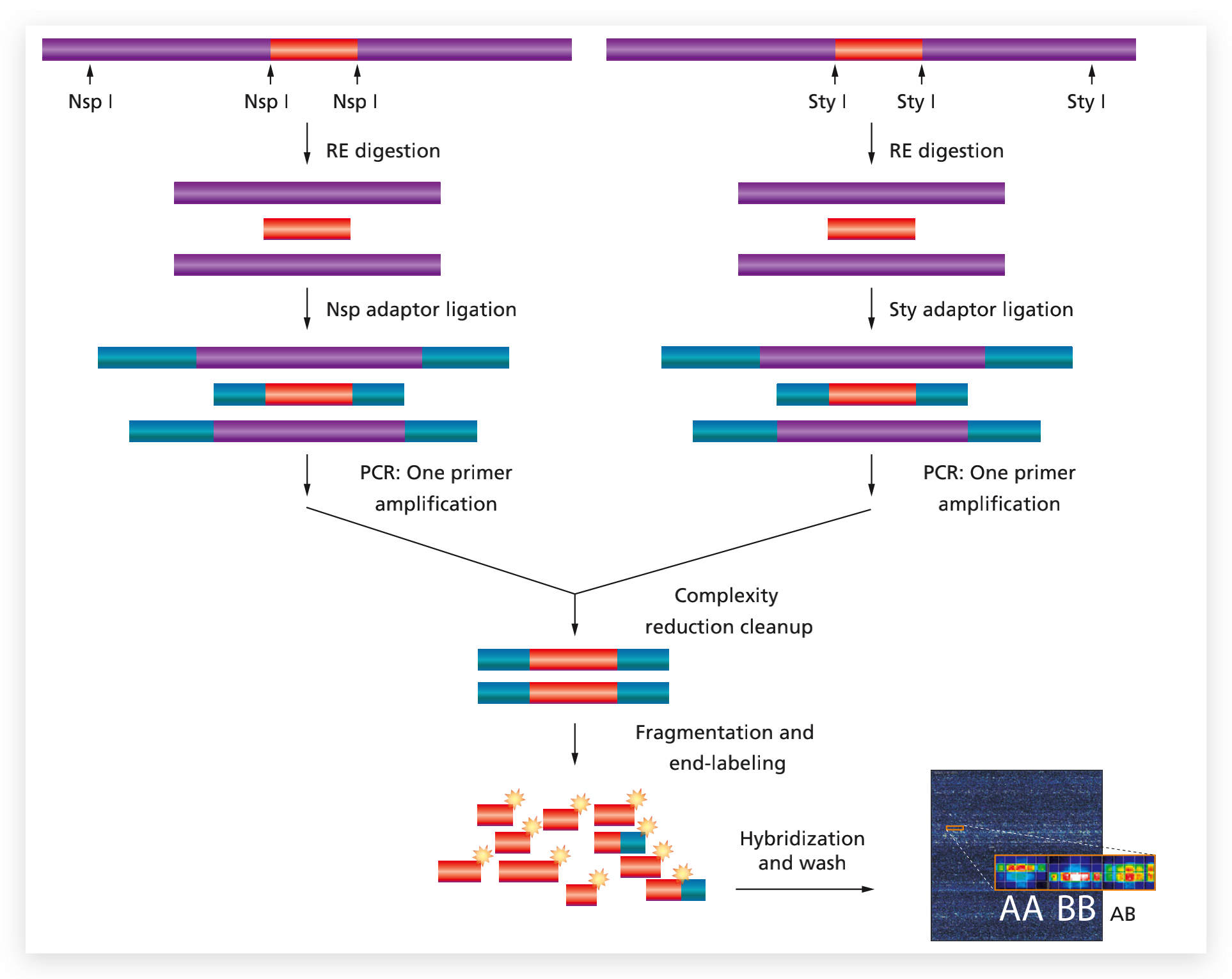
SNP array 5.0/6.0的检测数据来源
使用Affymetrix SNP 6.0 数据来定义重复的基因组区域以及该重复区域的拷贝数,这个pipeline使用已有的TCGA 第2水平的数据(normalized data) 和R包 DNAcopy 来实现CBS算法进行数据的分割。级别1的数据是原始的芯片强度数据,浏览了一下GDC上的数据,数据格式为CEL的,测序平台为SNP 6.0,这批原始数据是私密的。

级别1的数据
通常是用PICNIC等软件来处理原始的数据,得到分段记录的文件。TCGA级别2的Tangent Copy Number文件,通过对芯片强度数值进行归一化,估计原始的拷贝数,并且做切线归一化(tangent normalization)即减去在正常样本中发现的变异。Tangent Copy Number数据包含下面5列数据:
Chromosome Start End Num_Probes Segment_Mean
用DNAcopy包将TCGA级别2的 Tangent Copy Number 数据转换成 Copy Number Segment 数据,该文件以制表符分隔的格式将相邻染色体区域与log2比率段关联,与每个染色体区域相关联的带有强度值的探针的数量包括在该文件中(没有强度值的探针不包括在计数中),在拷贝数分割过程中,从男性中去除伪常染色体区探针集,从女性中去除Y染色体片段。这里还有一个 masked copy number segments ,使用的是和上述一样的方法,不同之处在于过滤步骤,去除了Y染色体和探针集中种系拷贝数变异,针对该数据使用GISTIC2来获得拷贝数变异数值文件Copy number Estimate,最后只保留编码蛋白质的基因,以及这些基因的拷贝数变异数值,噪音阈值设定在0.3,认为:
- CNV数值<-0.3则分类为删除事件(-1)
- CNV数值>0.3则分类为扩增事件(+1)
- -0.3<CNV数值<0.3则分类为中性事件(0)
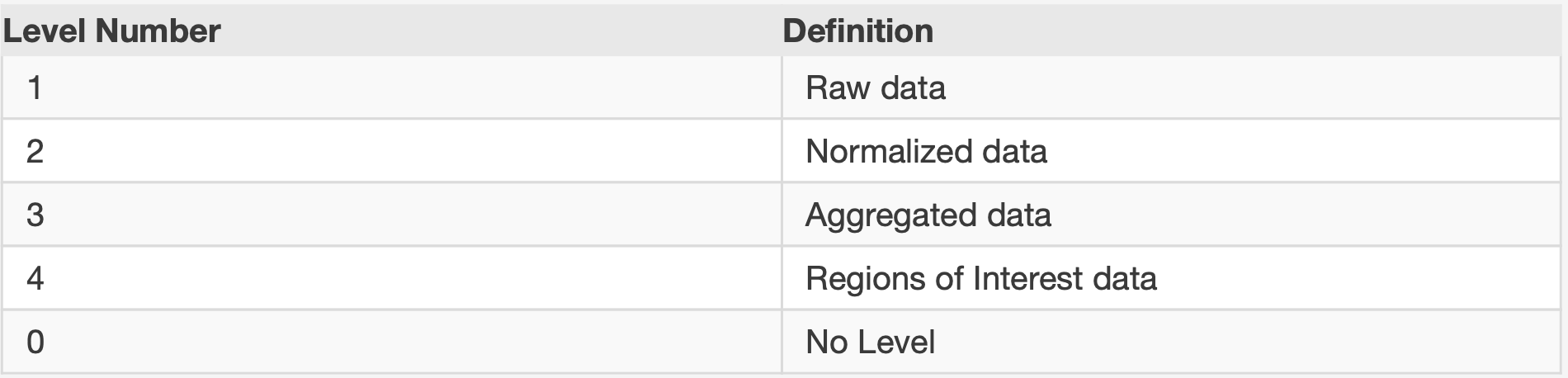
数据级别对应的数据类型
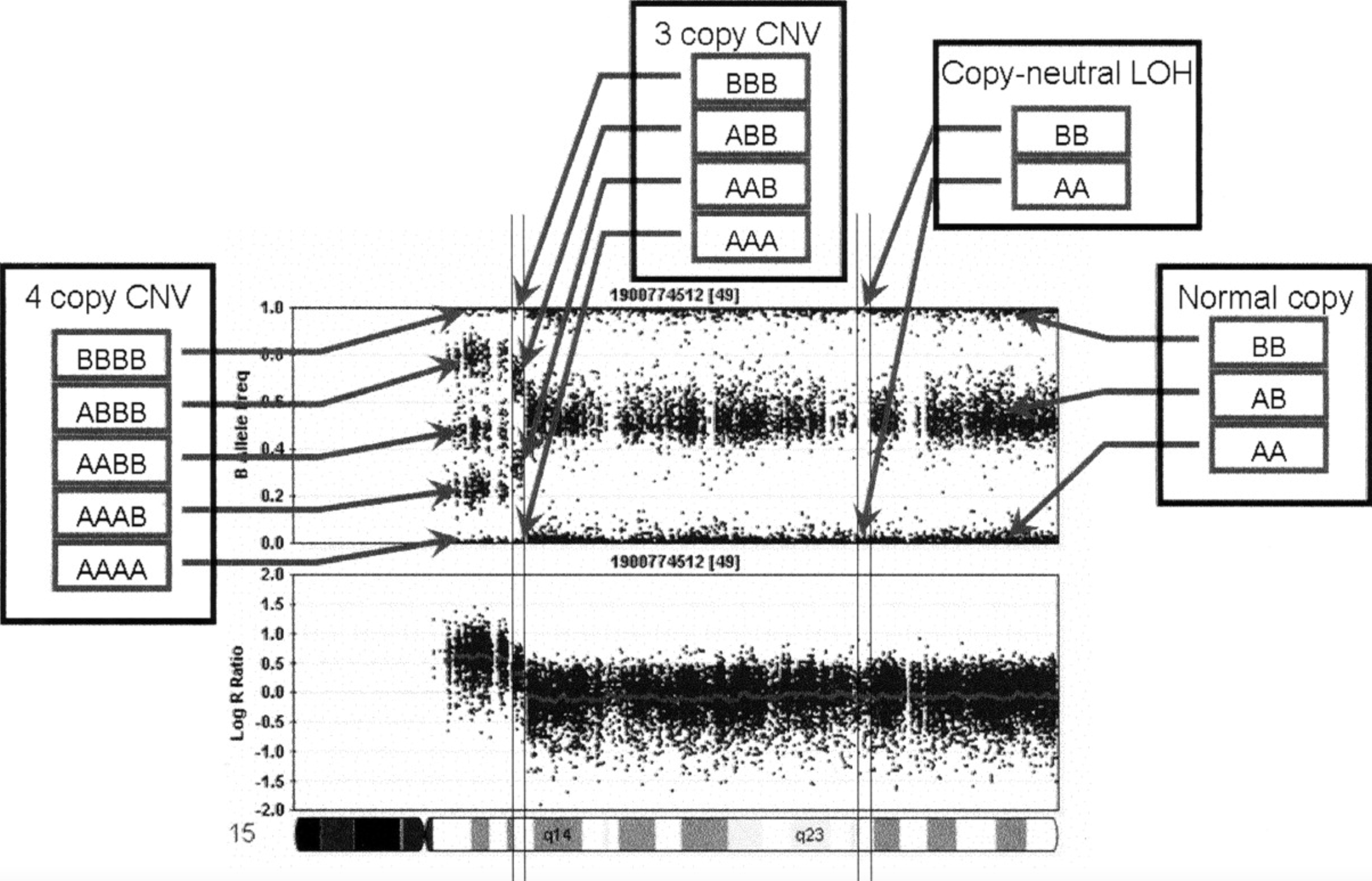
示例:个体15号染色体q臂的log R Ratio (LRR)和B Allele Freq (BAF) values
2.3.1 SNP芯片数据工具
-
PennCNV
-
DNAcopy
-
……
2.3.2 隐马尔可夫模型
2.4 全基因组(WGS)
全基因组的成本很高,针对全基因组CNV的检测,开发了一种CNV_seq的测序策略,指的是低深度全基因组测序,只需要5X的测序深度,就可以有效的检测CNV,全基因组CNV分析的算法/策略(以删除和新序列插入为例):
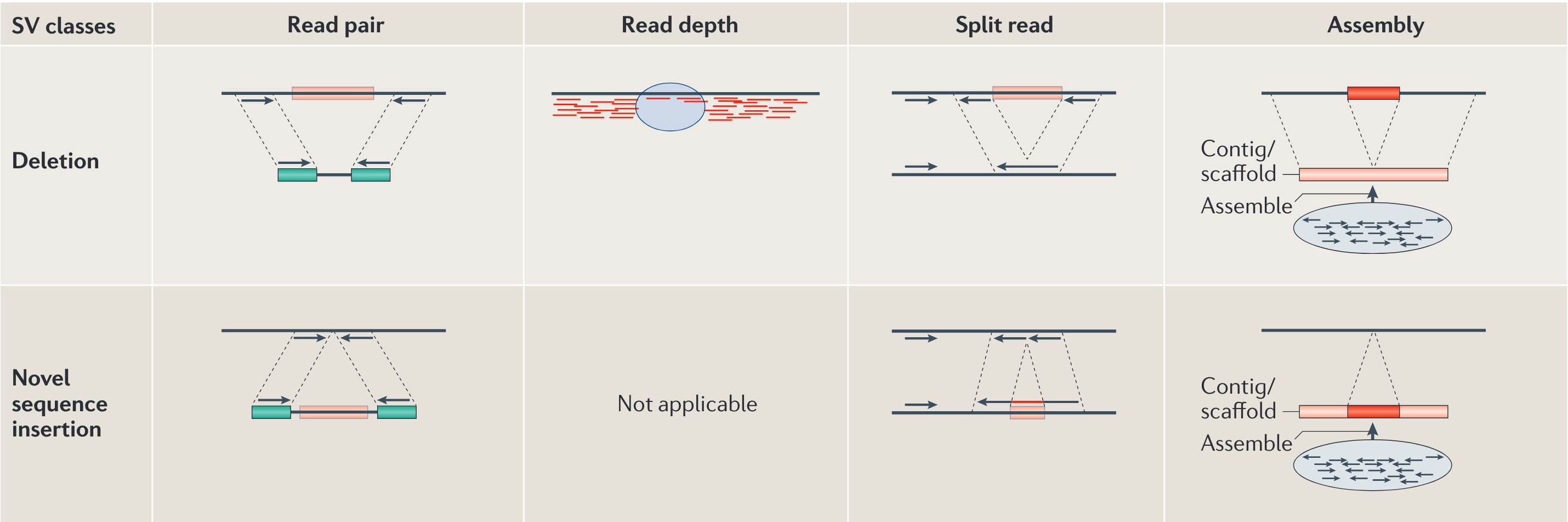
Genome structural variation discovery and genotyping
二代测序技术采用双端测序(图中绿色部分就是所测序列两端),得到双端的序列比对到参考基因组上,可以根据比对到参考基因组上的两端序列之间的间隔推测变异是否存在,大家都以参考基因组为基准,推断方法有四种以上主要的策略:
-
Read Pair(RP) : 是根据双端测序插入片段的长度分布来检测CNV的,以1为例,假设测得的基因序列是500bp的reads,但是匹配到参考基因组上发现,两端的序列匹配上之后序列比500bp还要长,那么认为两端序列之间发生删除后才是我们测到的序列。
软件工具:BreakDancer;PEMer;Ulysses
-
Read Depth(RD) : 通过测序堆在参考基因组上,某处reads很多堆在一起(可能是扩增),某处reads缺失(可能是插入),扩增的序列往往指在参考基因组上存在(或者相似度很高),而novel sequence insertion中novel意思是这段序列通常比较特殊,在参考基因组上找不到。
软件工具:CNVnator;ERDs
-
Split Read(SR) : 把reads mapping到基因组上,刚好reads被切开,切开的两端可以很好的mapping到参考基因组上。
软件工具:Pindel;SVseq2
-
Assmbly : 从头组装,不是和参考基因组去比对。通过测序得到的序列从头组装后得到的contig直接和参考基因组做序列比对,就可以知道发生变异的情况。
现在这四种方法都用,做得较好的软件:manta sv同时用到了这四种策略。
2.5 全外显子组(WES)
由于全基因组的成本挺高的,又考虑到外显子上的变异可能更具有致病性,因此可以基于WES做CNV分析,由于CNV区域的长度可能横跨了多个外显子或者基因,断点可能位于外显子以外的位置,所以PR,SR的策略无法应用到WES的CNV分析中,只能通过RD的策略进行分析。
可以使用CNVkit、XHMM来鉴定WES的CNV。
2.6 靶向测序
https://cloud.tencent.com/developer/article/1556103
http://www.njnad.com/marketing/421.html
https://cloud.tencent.com/developer/article/1556107
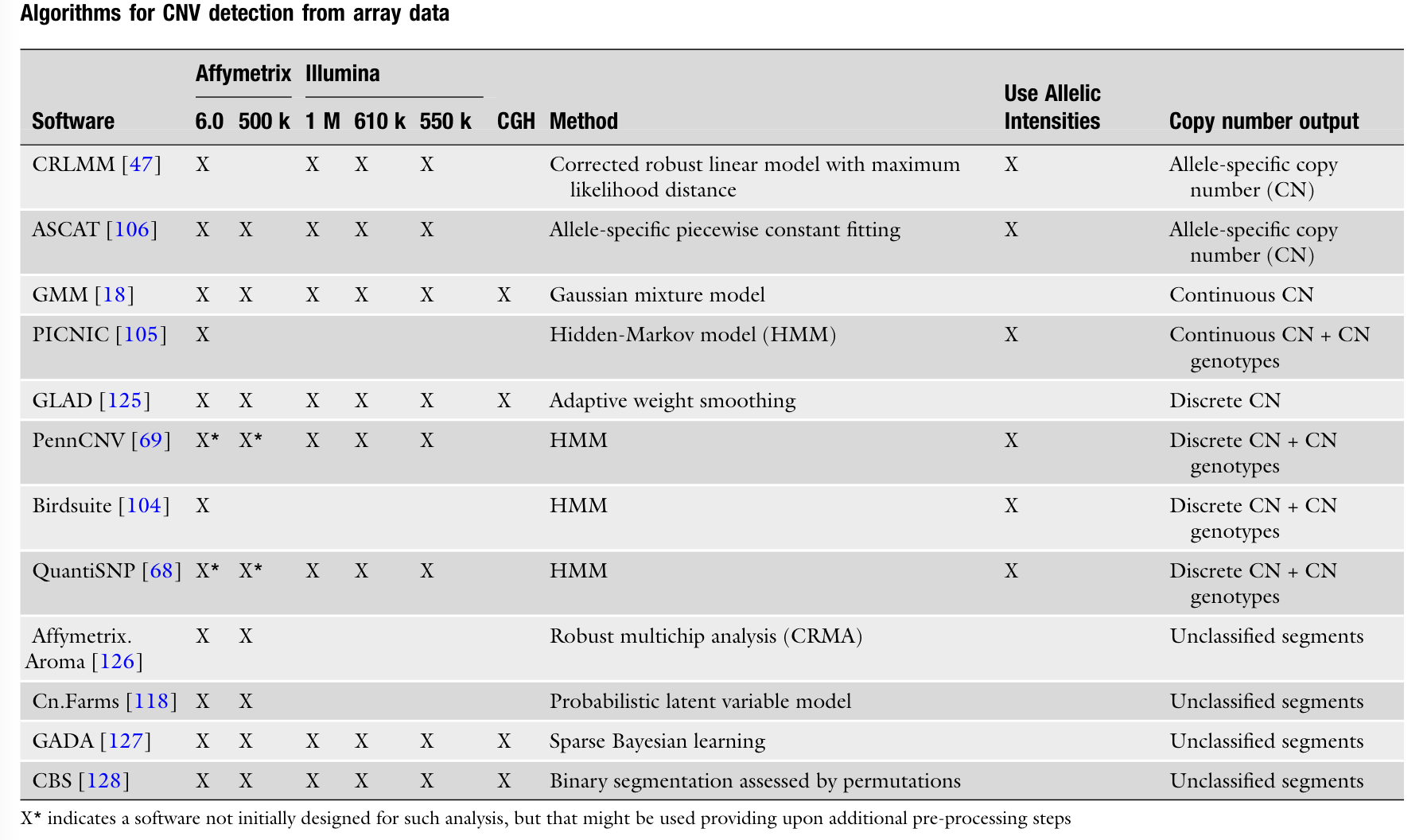
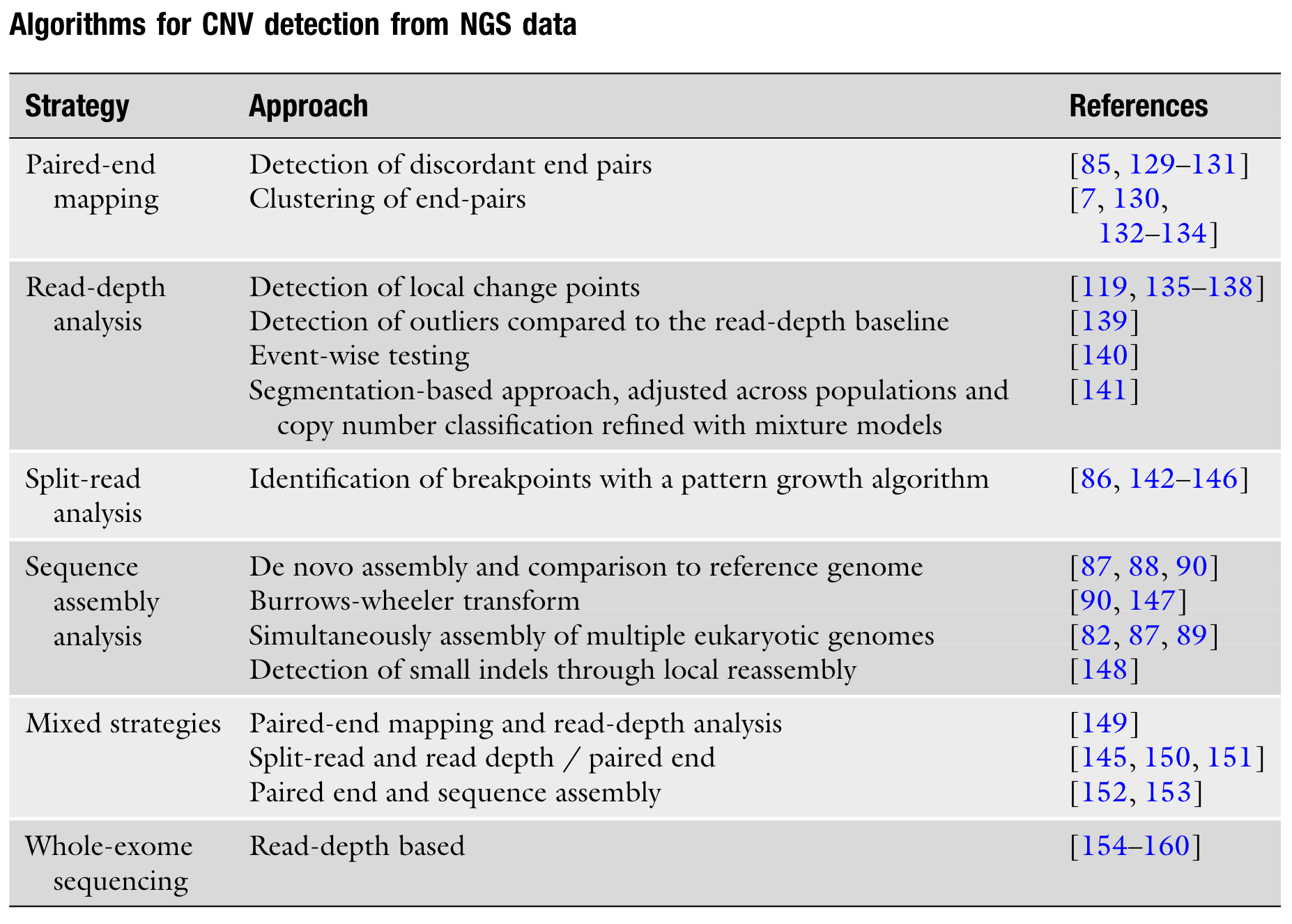
下期内容:
- 相对和绝对拷贝数数值的获取
- 示例:ABSOLUTE
- 使用隐马尔可夫模型检测CNV
- 主流获取CNV的工具比较
- 靶向测序检测CNV
- GATK所使用的拷贝数calling算法
- CBS算法除了判断断点,如何判断拷贝数数值?
- PennCNV能够同时判断断点和拷贝数数值咩?总强度用来判断拷贝数数值,相对强度BAF用来判断LOH咩?
- 我认为PennCNV有一个缺点就是,但是实际上可能拷贝数扩增不止4,那么其他的软件是怎么解决这个问题的?为了优化HMM的参数,使用Baum-Welch算法去优化,使用Vitebi算法来推断最可能的状态路径。排除了所有包含SNP小于等于2的CNV,因为这些CNV很肯定是假阳性的结果。
- 马尔可夫链依赖于初值的选择,初值的选择影响大吗?
- ……
参考资料
-
Kallioniemi, Anne, et al. “Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors.” Science 258.5083 (1992): 818-821.
-
Schena, Mark, et al. “Quantitative monitoring of gene expression patterns with a complementary DNA microarray.” Science 270.5235 (1995): 467-470.
-
Pinkel, Daniel, et al. “High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays.” Nature genetics 20.2 (1998): 207-211.
-
Alkan, Can, Bradley P. Coe, and Evan E. Eichler. “Genome structural variation discovery and genotyping.” Nature Reviews Genetics 12.5 (2011): 363-376.
-
Wang, Kai, et al. “PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data.” Genome research 17.11 (2007): 1665-1674.
-
图:Kai Wang et al. Genome Res. 2007;17:1665-1674
-
Circular Binary Segmentation from Jeremy Teibelbaum’s blog
-
CKVkit:https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1004873
-
隐马尔可夫模型:https://www.cnblogs.com/skyme/p/4651331.html